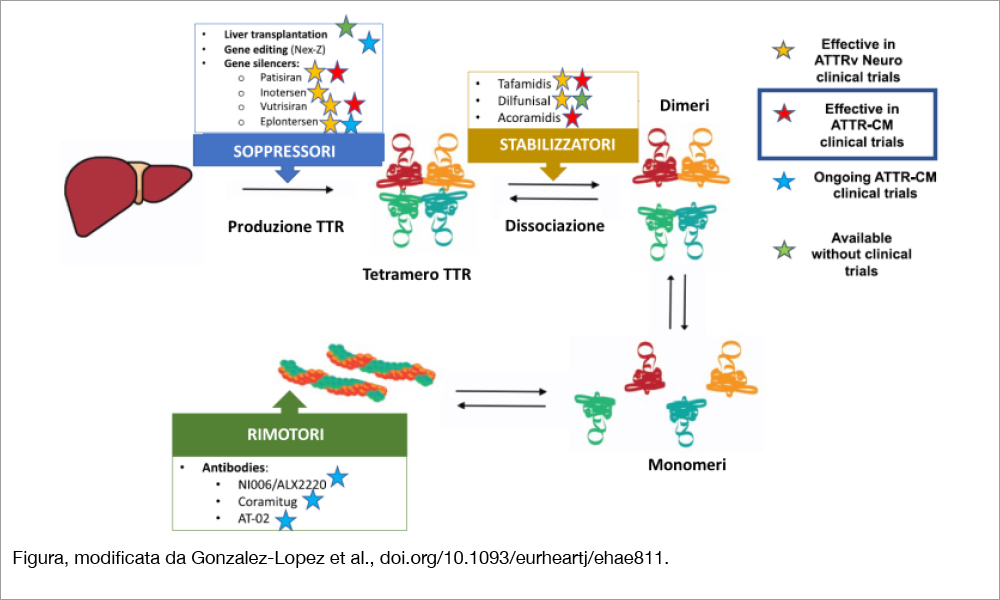
L’amiloidosi cardiaca da transtiretina (TTR) è una malattia progressiva causata dalla deposizione extracellulare di TTR a livello cardiaco. Può essere di origine ereditaria per variante genetica o, più frequentemente, senile correlata ad alterazione di meccanismi omeostatici (wild-type form -ATTRwt-). La cascata amiloidogenica è simile per le due forme: TTR è una proteina tetramerica prodotta dal fegato, che trasporta la tiroxina e la proteina legante la vitamina A. L’instabilità della molecola provoca dissociazione del tetramero in dimeri e monomeri con la produzione di aggregati insolubili di monomeri con alterato ripiegamento (misfolding) che generano fibrille di amiloide che si depositano[1]Subedi S, Sasidharan S, Nag N, Saudagar P, Tripathi T. Amyloid cross-seeding: mechanism, implication, and inhibition. Molecules 2022;27:1776. https://doi.org/10.3390/molecules27061776. L’amiloidosi TTR è una malattia sistemica, la cui prognosi, tuttavia, è soprattutto dipendente dalla alterazione cardiaca. Mentre un tempo si pensava fosse una patologia piuttosto rara, studi recenti, favoriti anche da mezzi diagnostici semplificati rispetto al passato, hanno mostrato come essa sia presente nel 12% dei pazienti con HFpEF, nel 10–15% degli anziani con stenosi aortica severa e nel 7% dei pazienti con spessore di parete ≥15 mm. L’armamentario terapeutico si è arricchito negli ultimi anni di nuovi farmaci. Essi possono essere divisi in:
- stabilizzatori, che limitano la frammentazione della molecola e la formazione di fibrille;
- soppressori della produzione di TTR da parte del fegato;
- rimotori, delle fibrille di amiloide dai tessuti (vedi Figura).
Tra i primi, tafamidis è ormai ampiamente utilizzato nella pratica clinica, in seguito ai dati favorevoli dello studio ATTR-ACT[2]Maurer MS, Schwartz JH, Gundapaneni B, et al. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med 2018;379:1007–16. https://doi.org/10.1056/NEJMoa1805689 ed è stato incorporato nelle Linee Guida del 2021con una raccomandazione di classe I nei pazienti in classe NYHA I e II. L’acoramidis è farmaco analogo, con ottimi risultati di efficacia (anche sulla mortalità) e safety, pur in una popolazione a rischio meno elevato rispetto a quella studiata con tafamidis, mostrati nello studio ATTRIBUTE-CM[3]Gillmore JD, Judge DP, Cappelli F, et al. Efficacy and safety of acoramidis in transthyretin amyloid cardiomyopathy. N Engl J Med 2024;390: 132–42. https://doi.org/10.1056/NEJMoa2305434.. I soppressori di TTR, approvati per la cura della polineuropatia, agiscono silenziando i geni responsabili per la produzione di TTR. Vutrisiran, studiato nel trial di fase III HELIOS-B, si è dimostrato efficace in una popolazione al 40% trattata anche con tafamidis. Non esistono studi di fase III per i rimotori di TTR, perciò non è possibile giudicarne efficacia e safety. La combinazione di farmaci con meccanismi d’azione diversa potrebbe avere effetti sinergici e migliorare la risposta clinica dei pazienti, ma non esistono al momento dimostrazioni cliniche in proposito.
Bibliografia
| ↑1 | Subedi S, Sasidharan S, Nag N, Saudagar P, Tripathi T. Amyloid cross-seeding: mechanism, implication, and inhibition. Molecules 2022;27:1776. https://doi.org/10.3390/molecules27061776 |
|---|---|
| ↑2 | Maurer MS, Schwartz JH, Gundapaneni B, et al. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med 2018;379:1007–16. https://doi.org/10.1056/NEJMoa1805689 |
| ↑3 | Gillmore JD, Judge DP, Cappelli F, et al. Efficacy and safety of acoramidis in transthyretin amyloid cardiomyopathy. N Engl J Med 2024;390: 132–42. https://doi.org/10.1056/NEJMoa2305434. |
Accedi per leggere tutto l'articolo
Inserisci i dati del tuo account su Cardiotalk per accedere e leggere tutto il contenuto dell'articolo.
Se non hai un account, clicca sul pulsante registrati e verrai reindirizzato al portale Cardiotalk per la registrazione.